Module Nine of CCE 281 Corrosion: Impact, Principles, and Practical Solutions
Factors Affecting Atmospheric Corrosion
The most important factor in atmospheric corrosion, overriding pollution or lack of it, is moisture, either in the form of rain, dew, condensation, or high relative humidity (RH). In the absence of moisture, most contaminants would have little or no corrosive effect.
Rain also may have a beneficial effect in washing away atmospheric pollutants that have settled on exposure surfaces. This effect has been particularly noticeable in marine atmospheres. On the other hand, if the rain collects in pockets or crevices, it may accelerate corrosion by supplying continued wetness in such areas as illustrated in the following Figure.

Galvanized bolting assembly after ten years of exposure to a deicing salt environment.
Dew and condensation are undesirable from a corrosion standpoint if not accompanied by frequent rain washing which dilutes or eliminates contamination. A film of dew, saturated with sea salt or acid sulfates, and acid chlorides of an industrial atmosphere provides an aggressive electrolyte for the promotion of corrosion. Also, in the humid Tropics where nightly condensation appears on many surfaces, the stagnant moisture film either becomes alkaline from reaction with metal surfaces, or picks up carbon dioxide and becomes aggressive as a dilute acid.
Temperature plays an important role in atmospheric corrosion in two ways. First, there is the normal increase in corrosion activity which can theoretically double for each ten-degree increase in temperature. Secondly, a little-recognized effect is the temperature lag of metallic objects, due to their heat capacity, behind changes in the ambient temperature.
As the ambient temperature drops during the evening, metallic surfaces tend to remain warmer than the humid air surrounding them and do not begin to collect condensation until some time after the dew point has been reached. As the temperature begins to rise in the surrounding air, the lagging temperature of the metal structures will tend to make them act as condensers, maintaining a film of moisture on their surfaces.
The period of wetness is often much longer than the time the ambient air is at or below the dew point and varies with the section thickness of the metal structure, air currents, RH, and direct radiation from the sun.
Cycling temperature has produced severe corrosion on metal objects in the Tropics, in unheated warehouses, and on metal tools or other objects stored in plastic bags. Since the dew point of an atmosphere indicates the equilibrium condition of condensation and evaporation from a surface, it is advisable to maintain the temperature some 10 to 15oC above the dew point to ensure that no corrosion will occur by condensation on a surface that could be colder than the ambient environment.
Relative humidity (RH)
RH is defined as the ratio of the quantity of water vapor present in the atmosphere to the saturation quantity at a given temperature, and it is expressed as %. A fundamental requirement for atmospheric corrosion processes is the presence of a thin film electrolyte that can form on metallic surfaces when exposed to a critical level of humidity. While this film is almost invisible, the corrosive contaminants it contains are known to reach relatively high concentrations, especially under conditions of alternate wetting and drying.
The critical humidity level is a variable that depends on the nature of the corroding material, the tendency of corrosion products and surface deposits to absorb moisture, and the presence of atmospheric pollutants. It has been shown that, for example, this critical humidity level is 60% for iron if the environment is free of pollutants. In the presence of thin film electrolytes, atmospheric corrosion proceeds by balanced anodic and cathodic reactions. The anodic oxidation reaction involves the corrosion attack of the metal, while the cathodic reaction is naturally the oxygen reduction reaction as shown in the following Figure.
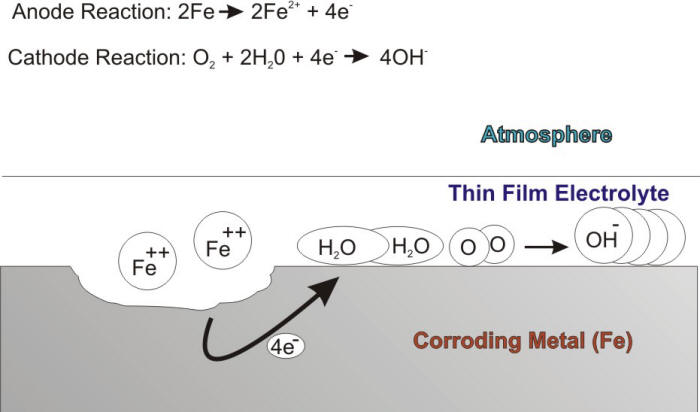
Schematic description of the atmospheric corrosion of iron.
Marine environments typically have high percent relative humidity (%RH), as well as salt rich aerosols. Studies have found that the thickness of the adsorbed layer of water on a zinc surface increases with %RH and that corrosion rates increase with the thickness of the adsorbed layer. There also seems to be a finite thickness to the water layer that, when exceeded, can limit the corrosion reaction due to limited oxygen diffusion. However, when metallic surfaces become contaminated with hygroscopic salts their surface can be wetted at lower %RH. The presence of magnesium chloride (MgCl2) on a metallic surface can make a surface apparently wet at 34% RH while sodium chloride (NaCl) on the same surface requires 77% RH to create the same effect.
The depression of the critical humidity levels on a metallic surface may seriously limit the use of some materials in various applications. Propose some design solutions to limit or avoid altogether such problems.
Calculate the dew point temperature for a RH of 45% when the ambient temperature is 22oC? ... when it is 26oC? You may have to consult the Internet or the textbook to solve this problem.
Deposition of aerosol particles
The behavior of aerosol particles in outdoor atmospheres can be explained by invoking the laws that govern their formation, movement and capture. These particles are present throughout the planetary boundary layer and their concentrations depend upon a multitude of factors including location, time of day or year, atmospheric conditions, presence of local sources, altitude and wind velocity. The highest concentrations are usually found in urban areas, reaching up to 108 and 109 particles per cm cube, with particle size ranging from around 100 µm to a few nm. Size is normally used to classify aerosol because it is the most readily measured property and other properties can be inferred from size information. The highest mass fraction of particles in an aerosol is characterized by particles having a diameter in the range of 8 µm to 80 µm. Some studies have also indicated that there is a strong correlation between wind speed and the deposition and capture of aerosols. In such a study of saline winds in Spain a very good correlation was found between chloride deposition rates and wind speeds above a threshold of 3 m s-1or 11 km h-1.
Aerosols can either be produced by ejection into the atmosphere, or by physical and chemical processes within the atmosphere (called primary and secondary aerosol production respectively). Examples of primary aerosols are sea spray and wind blown dust. Secondary aerosols are often produced by atmospheric gases reacting and condensing, or by cooling vapor condensation (gas to particle conversion). Once an aerosol is suspended in the atmosphere, it can be altered, removed or destroyed. It cannot stay in the atmosphere indefinitely, and average lifetimes are of the order of a few days to a week.
The lifetime of any particular particle depends on its size and location. Studies of the migration of aerosols inland of a sea coast have shown that typically the majority of the aerosol particles are deposited close to the shoreline (typically 400 to 600 m) and consist of large particles (>10µm diameter), which have a short residence time and are controlled primarily by gravitational forces . The aerosols that form also have mass and are subject to the influence of gravity, wind resistance, droplet dry-out, and possibilities of impingement on a solid surface, as they progress inland.
Pollutants
Sulfur dioxide (SO2), which is the gaseous product of the combustion of fuels that contain sulfur such as coal, diesel fuel, gasoline and natural gas, has been identified as one of the most important air pollutants which contribute to the corrosion of metals.
Less recognized as corrosion promoters, are the nitrogen oxides (NOx), which are also products of combustion. A major source of NOx in urban areas is the exhaust fumes from vehicles. Sulfur dioxide, NOx and airborne aerosol particles can react with moisture and UV lightto form new chemicals that can be transported as aerosols. A good example of this is the summertime haze over many large cities. Up to 50% of this haze is a combination of sulfuric and nitric acids.
| (previous) | Page 3 of 8 | (next) |
Connect with us
Contact us today